| The essentials: On July 24, 2026, the FDA approved Simtriyo (centanafadine, Otsuka Pharmaceutical) extended-release capsules for the treatment of ADHD in adults and pediatric patients aged 6 years and older weighing at least 20 kg (approximately 44 pounds). Simtriyo is the first and only approved norepinephrine, dopamine, and serotonin reuptake inhibitor (NDSRI) for any indication. This is a first-in-class mechanism that distinguishes centanafadine from every currently approved ADHD medication. What centanafadine is: an oral once-daily extended-release capsule that inhibits the reuptake of norepinephrine, dopamine, and serotonin simultaneously, increasing the availability of all three monoamines in the brain pathways involved in attention, executive function, and emotional regulation. Stimulants (amphetamines, methylphenidate) primarily increase dopamine and norepinephrine through reuptake inhibition and release. Atomoxetine (Strattera) is a selective norepinephrine reuptake inhibitor. Centanafadine is the first approved agent to add serotonin to the dopamine and norepinephrine target profile. The serotonin component is what differentiates Simtriyo from prior dual-mechanism ADHD drugs and is responsible for the emotional dysregulation and comorbid anxiety benefits observed in clinical data. The clinical basis: four Phase 3 trials across three age groups. Two adult trials (NCT03605680 and NCT03605836; n=859 total adults with ADHD aged 18 to 55): primary endpoint: change from baseline in AISRS (Adult ADHD Investigator Symptom Rating Scale) total score at week 6; both centanafadine 200 mg/day and 400 mg/day showed statistically significant and clinically meaningful improvements versus placebo; onset at week 1 (first post-baseline assessment); sustained through week 6. One adolescent trial (ages 13 to 17): primary endpoint: change from baseline in ADHD-RS-5 total score at week 6; high-dose group met primary endpoint with statistically significant improvement; benefit emerging by week 1. One pediatric trial (ages 6 to 12): primary endpoint: change from baseline in ADHD-RS-5 total score at week 6; high-dose group met primary endpoint with statistically significant improvement; benefit emerging by week 1. Low-dose groups did not reach statistical significance in pediatric or adolescent trials. Notable Phase 3b finding: in 315 adults with ADHD and comorbid anxiety (NCT06973577): AISRS improvement LS mean change minus 18.5 versus minus 12.6 for placebo; treatment difference minus 5.87; p less than 0.0001; HAM-A improvement (anxiety scale) also statistically significant (p=0.02); separation at week 1 maintained through week 8. Regulatory designations: Priority Review. DEA scheduling: Simtriyo is classified as a CNS stimulant and must receive a DEA Schedule classification before commercial launch; this standard process takes up to 3 months. How the DEA classifies it (Schedule II like amphetamines, or a lower schedule) will affect prescribing practices and commercial potential. Boxed warnings: suicidal ideation and behavior in children aged 6 to 12; and abuse, misuse, and addiction (with risk mitigation counseling on storage, disposal, and ongoing patient reassessment). Common adverse reactions: decreased appetite, nausea, rash, fatigue, upper abdominal pain, somnolence. Population: approximately 7 million children and 15.5 million adults in the United States have ADHD. |
|---|
Attention-deficit/hyperactivity disorder is the most commonly diagnosed neurodevelopmental condition in childhood and affects a substantial and often undertreated adult population. The FDA approved its first treatments for ADHD over 60 years ago. Since then, the pharmacological toolkit has evolved, but it has stayed within two broad categories: stimulants that drive up dopamine and norepinephrine in the prefrontal cortex, and non-stimulants that work more selectively on norepinephrine alone.
That framework has worked well for millions of patients. But it has also left clear gaps. The emotional dysregulation that characterizes many ADHD presentations, the hair-trigger frustration, the mood swings that follow a bad day, the inability to manage disappointment proportionally, these are not adequately addressed by dopamine and norepinephrine manipulation alone. The comorbid anxiety that co-occurs with ADHD in an estimated 50% of patients is poorly served by stimulants, which can exacerbate anxiety in many individuals. The 30% of patients who do not respond to or cannot tolerate existing medications continue cycling through options that were designed around the same two-target pharmacology.
Simtriyo (centanafadine, Otsuka) is the first drug for ADHD that adds serotonin to the target profile. Its three-neurotransmitter mechanism is not just a numbers upgrade. It is a qualitatively different pharmacological approach to a condition that has long been understood to involve more than two neurochemical systems.
The four Phase 3 trials, conducted across all ages from children to adults, showed consistent, statistically significant improvement in ADHD symptoms. A Phase 3b trial in adults with ADHD and comorbid anxiety showed that centanafadine improved both conditions simultaneously. And the drug demonstrated low abuse and dependence potential in clinical studies, which is a meaningful clinical consideration in a condition where stimulant diversion, misuse, and concerns about controlled substance prescribing remain persistent barriers to treatment access.
What ADHD Is: The Neurobiology and the Clinical Picture
Attention-deficit/hyperactivity disorder is a chronic neurodevelopmental disorder characterized by a persistent pattern of inattention, hyperactivity, and impulsivity that is inconsistent with the developmental level of the individual and interferes with functioning across settings. The DSM-5-TR recognizes three presentations: predominantly inattentive, predominantly hyperactive-impulsive, and combined presentation.
The prevalence of ADHD is substantial. In the United States, approximately 7 million children (approximately 9.4% of children aged 2 to 17 years) have a diagnosed ADHD, and approximately 15.5 million adults are currently estimated to have the condition. These numbers have grown with improved diagnostic criteria and increased awareness of adult ADHD, which was underrecognized for decades as a condition that was assumed to be outgrown in adolescence.
ADHD is highly heritable (heritability estimates of 70 to 80%) and is associated with dysregulation of the prefrontal cortex and its dopaminergic and noradrenergic inputs, which modulate attention, working memory, response inhibition, and executive function. Functional neuroimaging consistently shows reduced activation in fronto-striatal circuits in individuals with ADHD during tasks requiring sustained attention and executive control.
What standard neurobiology discussions of ADHD have historically underweighted is the role of serotonin. Serotonergic projections from the dorsal raphe nucleus reach the prefrontal cortex, striatum, and limbic system, where they modulate emotional reactivity, frustration tolerance, and mood stability, precisely the domains in which many individuals with ADHD experience their most functionally limiting symptoms. The emotional dysregulation of ADHD, which some researchers have proposed is a core feature rather than a comorbidity, is substantially serotonin-mediated. This is the gap that centanafadine’s third mechanism targets.
The ADHD Treatment Landscape and Where Simtriyo Fits
ADHD pharmacotherapy before Simtriyo was organized around two treatment classes:
Stimulants (Schedule II controlled substances)
Stimulants are the most prescribed and most effective first-line treatment for ADHD across all ages. They include:
Amphetamine-based stimulants: Amphetamine salts (Adderall, Adderall XR), lisdexamfetamine (Vyvanse), dextroamphetamine (Dexedrine). Mechanism: release of dopamine and norepinephrine from presynaptic terminals and inhibition of their reuptake.
Methylphenidate-based stimulants: Methylphenidate (Ritalin, Concerta, Daytrana, numerous others), dexmethylphenidate (Focalin). Mechanism: primarily reuptake inhibition of dopamine and norepinephrine without significant release.
Both classes produce robust improvements in attention, hyperactivity, and impulsivity in 70 to 80% of patients. Both are Schedule II controlled substances, requiring specific prescribing procedures, limiting the quantity dispensed, and restricting refills. Both carry abuse potential and are diverted for non-therapeutic use. Some patients cannot tolerate them due to cardiovascular effects, anxiety exacerbation, appetite suppression, or sleep disruption.
Non-stimulants
Atomoxetine (Strattera): A selective norepinephrine reuptake inhibitor, approved for ADHD in 2002 for children and adults. Takes 4 to 6 weeks to reach therapeutic effect. Effective for inattention; less effective than stimulants for hyperactivity in many patients. Not a controlled substance. Carries a boxed warning for suicidal ideation in pediatric patients.
Viloxazine ER (Qelbree): A selective norepinephrine reuptake inhibitor, approved 2021 for children and adolescents, 2022 for adults. Similar profile to atomoxetine but with some evidence of faster onset.
Alpha-2 adrenergic agonists (guanfacine ER/Intuniv; clonidine ER/Kapvay): Non-stimulant, non-controlled. Particularly useful for hyperactivity, impulsivity, and emotional dysregulation. Less effective for inattention as monotherapy. Often used as adjuncts to stimulants.
Where Simtriyo sits
Simtriyo occupies a novel position in this landscape. It is classified as a CNS stimulant (reflecting its dopaminergic and noradrenergic mechanism, which produces the physiological profile of a stimulant) but it is not a traditional amphetamine or methylphenidate. It adds serotonin reuptake inhibition to the dopamine and norepinephrine target profile. And it demonstrated low abuse and dependence potential in clinical studies, which is the basis for Otsuka’s expectation that the DEA may grant it a Schedule III or lower classification rather than the Schedule II designation that applies to amphetamines and methylphenidate.
The DEA scheduling decision will have significant commercial and clinical implications. A Schedule III or lower classification would allow phone-in and electronic prescriptions with refills (unlike Schedule II, which requires a new written or electronic prescription each month), which is a meaningful practical barrier reduction for patients on long-term ADHD therapy.
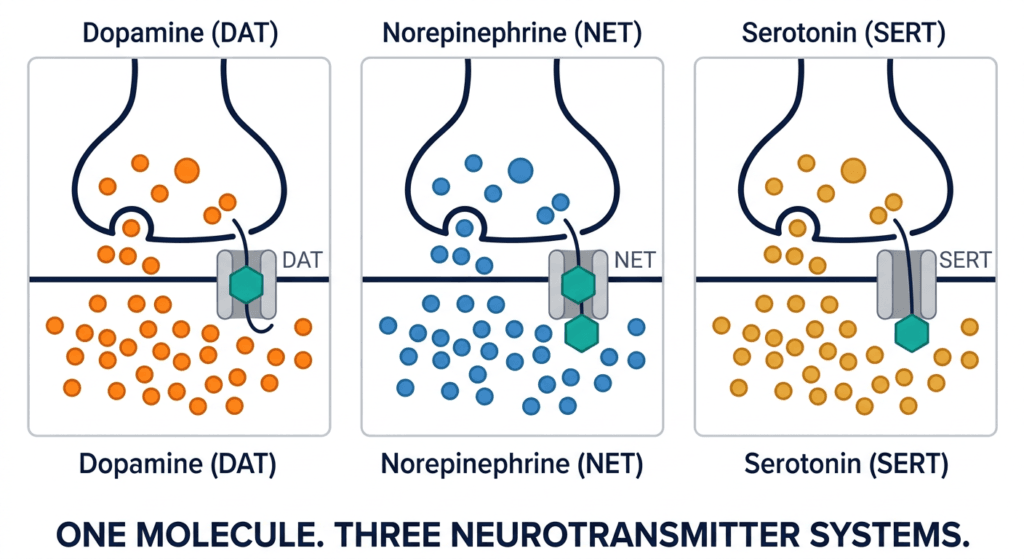
How Centanafadine Works: The Triple Reuptake Mechanism
Centanafadine inhibits the reuptake transporters for all three monoamine neurotransmitters: the norepinephrine transporter (NET), the dopamine transporter (DAT), and the serotonin transporter (SERT). By blocking these transporters, centanafadine prevents the reabsorption of norepinephrine, dopamine, and serotonin from the synaptic cleft back into the presynaptic neuron, increasing the availability of all three neurotransmitters in the synapse.
Dopamine transporter (DAT) inhibition: Increases synaptic dopamine in the prefrontal cortex and striatum, improving attention, motivation, and executive function. This is the mechanism shared with methylphenidate, though without the amphetamine-like release component.
Norepinephrine transporter (NET) inhibition: Increases synaptic norepinephrine in the prefrontal cortex, improving working memory, sustained attention, and response inhibition. This is the mechanism shared with atomoxetine and viloxazine, as well as with amphetamines.
Serotonin transporter (SERT) inhibition: Increases synaptic serotonin in the prefrontal cortex, limbic system, and striatum. Serotonin modulates emotional reactivity, frustration tolerance, anxiety, and mood stability. This is the mechanism shared with SSRIs and SNRIs, but in a drug designed primarily for ADHD rather than depression or anxiety.
The combination of all three mechanisms in a single molecule produces a pharmacological effect that simultaneously addresses the attention and executive function deficits of ADHD (via dopamine and norepinephrine) and the emotional dysregulation and comorbid anxiety that frequently accompany them (via serotonin). As Dr. Greg Mattingly noted in a discussion of centanafadine’s mechanism: the serotonin component was shown to help anxiety and mood in people with ADHD, since the drug kind of has an SSRI built in.
The extended-release capsule formulation delivers the drug over the course of the day, enabling once-daily dosing that maintains therapeutic concentrations through school or work hours without the rebound effects that can accompany immediate-release formulations.
The Four Phase 3 Trials: What the Data Shows
The FDA based its approval on the totality of evidence from four Phase 3 trials, spanning adults, adolescents, and children, with consistent findings of statistically significant and clinically meaningful ADHD symptom improvement in the higher-dose groups.
Adult trials: NCT03605680 and NCT03605836
Two randomized, double-blind, placebo-controlled Phase 3 trials in 859 adults aged 18 to 55 years with ADHD evaluated centanafadine sustained-release at 200 mg/day or 400 mg/day (administered in divided twice-daily doses) versus placebo over 6 weeks. The primary endpoint was the change from baseline in the AISRS (Adult ADHD Investigator Symptom Rating Scale) total score at week 6.
Both centanafadine doses (200 mg/day and 400 mg/day) demonstrated statistically significant and clinically meaningful improvement in AISRS total score versus placebo at week 6. Statistical separation from placebo emerged as early as week 1, the first post-baseline assessment time point, and was maintained through the 6-week study period.
Post-hoc analyses of the 744-patient adult dataset showed improvements in patient-reported executive function, including time management, planning and prioritization, task initiation and completion, and working memory. Improvements in emotional regulation measures, including reduced mood shifts, emotional overactivity, and anger outbursts, were also observed in the same post-hoc analyses.
Adolescent trial (ages 13 to 17)
A 6-week, randomized, double-blind, placebo-controlled, fixed-dose trial with weight-based dosing evaluated centanafadine extended-release in adolescents aged 13 to 17 years. The primary endpoint was change from baseline in ADHD-RS-5 (ADHD Rating Scale, fifth edition) total score at week 6. The high-dose group met the primary endpoint with statistically significant improvement versus placebo. Benefit was observed as early as week 1. The low-dose group did not reach statistical significance.
Pediatric trial (ages 6 to 12)
A 6-week, randomized, double-blind, placebo-controlled, fixed-dose trial with weight-based dosing evaluated centanafadine extended-release in children aged 6 to 12 years. The primary endpoint was change from baseline in ADHD-RS-5 total score at week 6. The high-dose group met the primary endpoint with statistically significant improvement versus placebo. Benefit was observed as early as week 1. The low-dose group did not reach statistical significance.
The weight minimum of 20 kg (approximately 44 pounds) for the approved indication reflects both the weight-based dosing approach in the pediatric trials and the pharmacokinetic considerations for centanafadine at the approved doses in the youngest and smallest children.
Phase 3b trial in ADHD with comorbid anxiety (NCT06973577)
Beyond the core NDA-supporting trials, Otsuka conducted a Phase 3b study in 315 adults aged 18 to 65 years with ADHD and comorbid anxiety (generalized anxiety disorder and/or social anxiety disorder) to evaluate centanafadine XR 280 mg once daily over 8 weeks.
| Endpoint | Centanafadine 280 mg (n=approximately 158) | Placebo (n=approximately 157) | Result |
|---|---|---|---|
| AISRS change from baseline at week 8 (primary) | LS mean minus 18.5 | LS mean minus 12.6 | Treatment difference minus 5.87; p less than 0.0001 |
| HAM-A change from baseline at week 8 (key secondary) | LS mean minus 12.5 | LS mean minus 10.6 | p=0.02 |
| Statistical separation | Week 1 (first post-baseline) | — | Maintained through week 8 |
Source: Otsuka Phase 3b press release. June 25, 2026. NCT06973577.
The simultaneous improvement in both ADHD and anxiety symptoms from a single drug in a population that traditionally requires two medications addresses one of the most clinically significant gaps in ADHD management. The 50% of ADHD patients who have comorbid anxiety are often undertreated for one or both conditions because stimulants can exacerbate anxiety, and adding an SSRI or SNRI to a stimulant regimen increases medication burden and interaction risk. A single drug that addresses both represents a meaningful clinical advance for this population.
Safety: What the Prescribing Information Covers
Boxed warnings
Simtriyo carries two boxed warnings, the FDA’s strongest safety designation:
Suicidal ideation and behavior in children aged 6 to 12: Centanafadine carries a boxed warning for suicidal ideation and behavior specifically in the 6-to-12 age group. This warning is supported by data from the pediatric trials and reflects the same class-level concern that applies to other ADHD treatments including atomoxetine and viloxazine. Caregivers should monitor children for new or worsening depression, unusual changes in behavior, or suicidal thoughts, particularly early in treatment or after dose changes. Clinical monitoring with frequent follow-up visits is recommended during initial titration in this age group.
Abuse, misuse, and addiction: As a CNS stimulant with dopaminergic mechanism, centanafadine carries an abuse potential warning. The prescribing information requires counseling patients and caregivers on the risk of abuse and misuse, safe storage (keeping the medication secure and away from others), proper disposal of unused medication, and ongoing reassessment of the patient’s need for the drug at each visit. Centanafadine demonstrated low abuse and dependence potential in clinical studies, which is factored into the pending DEA scheduling decision.
Common adverse reactions
The most common adverse reactions reported in ADHD clinical trials, occurring at higher rates than placebo, include: decreased appetite, nausea, rash, fatigue, upper abdominal pain, and somnolence. This adverse reaction profile is broadly consistent with CNS stimulant class effects, though the absence of headache from the list and the presence of rash distinguish the centanafadine profile somewhat from amphetamine formulations.
Key warnings and precautions
Cardiovascular monitoring: As with all CNS stimulants, centanafadine may affect heart rate and blood pressure. Cardiovascular status should be evaluated before initiating and monitored during treatment. Centanafadine should be used with caution in patients with pre-existing cardiac conditions, and the prescribing information should be reviewed for specific guidance.
Psychiatric adverse effects: Beyond the suicidality boxed warning in young children, psychiatric adverse effects including irritability, mood changes, and psychotic or manic episodes have been reported with stimulant class medications. Monitor for emergence of psychiatric symptoms during treatment.
Growth monitoring in children: As with other stimulant medications, height and weight monitoring is appropriate in pediatric patients on long-term centanafadine therapy.
The DEA Scheduling Question: A Critical Pre-Commercial Consideration
Before Simtriyo can be commercially dispensed in the United States, the Drug Enforcement Administration must assign it a controlled substance schedule. This is a standard regulatory process for all drugs with CNS activity and abuse potential, and it can take up to three months from FDA approval.
The question of which schedule is genuinely important:
A Schedule II designation (the classification of amphetamines and methylphenidate) would impose: no telephone or electronic prescriptions with refills; patients must obtain a new written or electronic prescription each month; some states impose additional restrictions. This schedule reflects the highest abuse potential among clinical controlled substances.
A Schedule III, IV, or V designation would allow: prescriptions with refills; electronic transmission; fewer administrative burdens per refill. Schedule III is where buprenorphine for opioid use disorder and many other abusable but non-amphetamine drugs sit.
Centanafadine’s clinical trial data showing low abuse potential, and its structural and pharmacological differentiation from classical amphetamines, support Otsuka’s expectation that the DEA may assign a lower schedule than amphetamines. Jefferies analysts noted that a lower schedule could give Simtriyo a competitive edge in a crowded ADHD market, and that how the DEA classifies it could determine its commercial potential.
What This Means for Clinicians and Patients
For psychiatrists, pediatricians, and primary care providers
Simtriyo introduces a genuinely new pharmacological option in the ADHD landscape. The triple reuptake mechanism is not a repackaging of existing approaches. It provides a new tool for the following patient profiles:
Patients with ADHD and comorbid anxiety, who cannot tolerate stimulants or who have had anxiety exacerbated by them. The Phase 3b comorbid anxiety data showing simultaneous improvement in both ADHD and anxiety symptoms from one drug is a clinically important finding.
Patients with prominent emotional dysregulation, the impulsive anger, frustration intolerance, and mood lability that sit at the intersection of ADHD and emotional disorders and are poorly addressed by traditional stimulants.
Patients who have not responded to or cannot tolerate existing stimulants or non-stimulants. The additional mechanistic target (serotonin) may produce therapeutic benefit in patients where dopamine/norepinephrine-focused agents have been insufficient.
Patients or families concerned about controlled substance prescribing, if the DEA assigns a lower schedule than amphetamines.
The DEA scheduling outcome, expected within approximately three months of the July 24 approval, will be an important milestone that determines the practical prescribing workflow. Most clinicians will want to wait for this clarification before integrating Simtriyo into their prescribing routines.
For patients and families
ADHD is a highly individualized condition. Not every patient will respond the same way to any given medication, and finding the right ADHD treatment often involves trial and adjustment of medications, doses, and formulations over time. Simtriyo adds a new first-in-class option to that process, with a mechanism specifically designed to address the emotional and anxiety-related dimensions of ADHD that current medications often leave inadequately treated.
As Dr. Lenard Adler, director of the adult ADHD program at NYU Langone Health and an investigator on the centanafadine trials, noted: having more therapeutic choices is important because ADHD is a highly individualized condition and treatment decisions should reflect the unique needs of each patient.
For families navigating a pediatric ADHD diagnosis, the weight requirement of at least 20 kg (approximately 44 pounds) should be confirmed before treatment initiation. The suicidal ideation boxed warning in children aged 6 to 12 is a prescribing information requirement that warrants close communication with the prescribing clinician about monitoring, visit frequency in early treatment, and what behavioral changes to watch for and report.
The Attention Deficit Disorder Association (ADDA) (add.org) and CHADD (Children and Adults with ADHD) (chadd.org; 1-800-233-4050) maintain current treatment information, prescriber directories, and patient community resources for adults and families navigating ADHD management.
Sources
FDA approval announcement: FDA approves centanafadine extended-release capsules for ADHD. FDA.gov. July 24, 2026. Full announcement.
Otsuka FDA approval press release: Otsuka receives FDA approval for first-in-class SIMTRIYO (centanafadine) for the treatment of attention-deficit hyperactivity disorder (ADHD) in adults and pediatric patients aged 6 years and older. Otsuka. July 24, 2026.
Drugs.com approval news: FDA Approves Simtriyo (centanafadine) for the Treatment of Attention-Deficit Hyperactivity Disorder (ADHD). drugs.com. July 24, 2026.
Psychiatric Times (first-in-class NDSRI mechanism, four Phase 3 trials, executive function post-hoc): FDA Approves Centanafadine for ADHD in Children, Adolescents, and Adults. psychiatrictimes.com. July 2026. Full article.
Psychiatric Times (clinical development path, AISRS primary endpoint, adult and pediatric trial design): Centanafadine’s Route to FDA Review: A Clinical Path of the ADHD Medication. psychiatrictimes.com.
Managed Healthcare Executive (AISRS week 1 onset, boxed warning detail, DEA scheduling context): FDA approves Simtriyo, the first triple reuptake inhibitor for ADHD. managedhealthcareexecutive.com. July 2026. Full article.
Fierce Pharma (DEA schedule, commercial analysis, Jefferies note, Dr. Adler quote): FDA approves Otsuka’s first-in-class ADHD drug Simtriyo. fiercepharma.com. July 2026.
Drug Topics (adult n=744 post-hoc data, pediatric/adolescent high-dose-only result, boxed warnings, week 1 onset): FDA Approves First-in-Class Centanafadine for Treatment of ADHD. drugtopics.com. July 2026.
ADDitude Magazine (serotonin-SSRI analogy quote, FDA approval context, 280 mg anxiety trial): Centanafadine (Simtriyo) Receives FDA Approval as ADHD Medication. additudemag.com.
ADDitude clinical path article (Phase 2 dose, pediatric trial design, low-dose negative results, Dr. Wilens investigator reference): Centanafadine: Forthcoming Triple Reuptake Inhibitor ADHD Drug. additudemag.com.
Otsuka Phase 3b comorbid anxiety press release (exact LS mean data, p-values): Otsuka Announces Positive Phase 3b Results for Centanafadine in Adults with ADHD and Comorbid Anxiety. otsuka-us.com. June 25, 2026.
Pharmacy Times (AISRS LS mean data, HAM-A secondary endpoint, pediatric trial detail): Centanafadine Demonstrates Statistically Significant, Clinically Relevant Improvements in ADHD Symptoms. pharmacytimes.com.
Otsuka ASCP 2026 post-hoc press release (executive function, emotional dysregulation domains from n=744): Otsuka Presents New Phase 3 Post Hoc Analyses of Centanafadine at ASCP 2026. otsuka-us.com. May 2026.
Medscape (first NDSRI approved, prevalence statistics): FDA Approves First NDSRI for ADHD in Adults and Children. medscape.com. July 2026.
OncoDaily / OncoLibrary (mechanism overview, 20 kg weight requirement): FDA Approves Zidesamtinib (Jideytro) for ROS1-Positive NSCLC. oncodaily.com.
Adult Phase 3 trial registrations: NCT03605680 and NCT03605836. ClinicalTrials.gov.
Phase 3b comorbid anxiety trial registration: NCT06973577. ClinicalTrials.gov.
ADHD overview: Attention Deficit Hyperactivity Disorder. StatPearls. NCBI.
Simtriyo prescribing information: SIMTRIYO (centanafadine) Extended-Release Capsules Prescribing Information. Otsuka. 2026.
Simtriyo approval history: Simtriyo FDA Approval History. drugs.com.
Patient resources: CHADD (Children and Adults with ADHD): 1-800-233-4050 | Attention Deficit Disorder Association (ADDA) | Otsuka Simtriyo patient support | CDC ADHD information
| Disclaimer: Health Evidence Digest provides general information about FDA approvals and health research for educational purposes. This content is not a substitute for professional medical advice. Simtriyo (centanafadine) carries a boxed warning for suicidal ideation and behavior in children aged 6 to 12 years, and a boxed warning for abuse, misuse, and addiction. Commercial availability awaits DEA scheduling, which is expected within approximately three months of FDA approval. Treatment decisions for ADHD, including the selection and initiation of any pharmacologic therapy, should be made in close collaboration with a board-certified psychiatrist, pediatrician, or primary care provider experienced in ADHD management, taking into account the individual patient’s clinical profile, comorbidities, and treatment history. |
|---|